Crime contre l'humanité
Vaccination des nourrissons : l’alerte rouge
Sans surprise, la FDA vient d’autoriser la vaccination contre le COVID des nourrissons et des enfants âgés entre 6 mois et 5 ans. Trois jours plus tôt, un comité d’experts s’était prononcé à l’unanimité, en faveur de cette extension. La généticienne Alexandra Henrion-Caude alerte aujourd’hui en signalant l’existence de 58 cas de bébés de moins de 3 ans injectés ayant subi des effets indésirables graves, dont certains mortels, alors que la FDA a conclu à l’absence d’effets indésirables chez les tout-petits.

L’avis rendu le 15 juin 2022 par le Comité consultatif sur les vaccins et les produits biologiques connexes (VRBPAC) et confirmé hier par la FDA concerne les deux vaccins Pfizer et Moderna. Plusieurs États avaient déjà anticipé cette décision et passé commande de millions de doses. Elles pourront donc être administrées dès le 21 juin aux enfants dès l’âge de 6 mois et jusqu’à 5 ans pour Pfizer (schéma vaccinal à deux doses) ou 6 ans pour Moderna (schéma à trois doses), bien que celui-ci soit interdit dans de nombreux pays pour les adultes de moins de 30 ans en raison de sa toxicité cardiaque.
C’est donc un nouveau pas qui est franchi dans l’horreur, mais qui résume à lui seul l’abjection qu’est la campagne de vaccination contre le COVID. Les conclusions de l’essai clinique sur lesquelles se sont basés les experts pour recommander la vaccination des tout-petits révèlent en effet une accumulation de fraudes et de signaux hautement inquiétants, incompatibles avec une homologation qui ouvre potentiellement la porte d’une future obligation vaccinale. Les données de pharmacovigilance révélées par la généticienne Alexandra Henrion-Caude complètent un tableau alarmant que nous détaillons ici.
L’extension de la vaccination enfreint la législation encadrant les autorisations d’utilisation d’urgence
L’association Children’s Health Defense, portée par l’avocat Robert F. Kennedy, Jr, a recensé les éléments qui auraient dû conduire à un rejet uninime de la demande des laboratoires par le VRBPAC, comme l’y exhortaient de nombreux experts, victimes d’effets secondaires et décideurs politiques.
Début juin, les membres du Congrès avaient en effet soumis au commissaire de la FDA une liste de 13 questions mettant en cause la sécurité et l’efficacité des injections pour les tout-petits, dans une lettre qui n’a à jour obtenu aucune réponse.
Ces éléments ont été portés à la connaissance du comité consultatif de la FDA lors de l’audience du 15 juin. C’est donc sur la base de ces informations que celui-ci s’est prononcé en faveur de l’inoculation des nourrissons et des jeunes enfants.
Plusieurs intervenants ont pourtant soulevé le fait qu’une telle extension de la vaccination constituerait une violation des règles d’octroi d’une autorisation d’utilisation d’urgence (Emergency Use Authorization, EUA) dont elle bafouerait deux conditions essentielles.
L’efficacité alléguée par les laboratoires eux-mêmes n’atteint pas le seuil réglementaire de 50 % fixé par la législation
L’efficacité du vaccin Pfizer (80%) a été calculée 30 jours après la 3e dose, soit avant son prévisible effondrement, et obtenue en combinant toutes les tranches d’âge. Celle du vaccin Moderna serait respectivement de 51% environ contre l’infection par Omicron chez les enfants de moins de 2 ans, versus 43,7% déclarés en mars par le laboratoire dans un communiqué de presse, et de 37 % chez les enfants de 2 à 5 ans, avec la quasi-certitude d’un booster à injecter également :
« Les directives traitent également de l’importance de veiller à ce que la taille des essais cliniques soit suffisamment grande pour démontrer l’innocuité et l’efficacité d’un vaccin. Il indique que la FDA s’attendrait à ce qu’un vaccin COVID-19 prévienne la maladie ou diminue sa gravité chez au moins 50% des personnes vaccinées. »
FDA. Coronavirus (COVID-19) Update: FDA Takes Action to Help Facilitate Timely Development of Safe, Effective COVID-19 Vaccines. 2020 June 30.
Cette infraction est parfaitement assumée par la FDA puisque le directeur du Centre d’évaluation et de recherche sur les produits biologiques de l’agence, le Dr. Peter Marks, avait déclaré le mois dernier devant le Congrès américain que « le régulateur ne refuserait pas l’autorisation d’un vaccin pédiatrique s’il ne respectait pas le seuil d’efficacité de 50% de l’agence pour bloquer les infections symptomatiques », selon The Defender. Il expliquait ailleurs pourquoi.
En d’autres termes, la FDA n’a jamais eu d’illusion sur l’inefficacité du vaccin, elle a juste assuré aux laboratoires les conditions pour qu’ils puissent s’affranchir de cette exigence pourtant cruciale.
La balance bénéfice-risque des vaccins est incompatible avec leur homologation
Une autorisation d’urgence ne peut être accordée qu’à la triple condition que cette urgence « présente un risque de décès pour le groupe cible, que le produit soit efficace pour prévenir la maladie, qu’il soit sûr et que les avantages doivent l’emporter sur le risque ». Or aucune de ces conditions n’est remplie pour les tout-petits, selon l’avocate Kathlyn Hinesley qui a rappelé lors de l’audience trois faits majeurs concernant l’efficacité et la sécurité des injections :
- le taux de survie pour les enfants sans comorbidités qui contractent le COVID-19 est de 99,98%. Le taux de létalité (risque de mourir du COVID en cas d’infection) au sein de cette tranche d’âge est donc de 0,02 %. Compte tenu du risque de contamination, le taux de mortalité (risque absolu de mourir du COVID) s’élèverait alors à 0,00001487671 chez les enfants et à 1 sur un million chez les moins de 5 ans ;
- les vaccins contre le COVID-19 sont associés à une augmentation de 38% des cas de COVID et à une augmentation de 31% des décès selon une étude d’envergure conduite sur 145 pays (Beatie KA) ;
- le nombre d’événements indésirables touchant les enfants âgés de 5 à 17 ans recensés dans la base de pharmacovigilance américaine (VAERS) incluait, au 3/06/2022, 8 811 effets indésirables graves, dont 114 décès et 1 346 cas de myocardite potentiellement mortelle.
Comment ces effets secondaires seront-ils diagnostiqués chez des nourrissons qui n’ont pas la capacité de s’exprimer et de décrire leurs symptômes ? Ils ne le seront sans doute pas, pas vu pas pris. D’où la conclusion de Kathlyn Hinesley :
« Nous pouvons supposer que si ces vaccins sont autorisés, certains bébés mourront. Les avantages de ces vaccins sont discutables et les risques sont clairs. »
Kathlyn Hinesley
Les méthodes de la FDA démontrent un recul dramatique de l’éthique de l’agence
Lors de l’audience ont également été signalés plusieurs points incriminant l’éthique dont l’agence a fait preuve depuis le début de la pandémie pour approuver les précédentes autorisations d’urgence :
- l’absence de prise en compte par la FDA de « signaux de sécurité massifs », concernant notamment des pathologies gravissimes comme les maladies à prions, les cancers ou des problèmes d’infertilité, aujourd’hui étayés par de nombreuses études, confirmés les données de la pharmacovigilance et cohérents avec la présence de nanoparticules lipidiques dans les vaccins.
Le SM-102 de Moderna est par exemple associé à un risque accru de cancer, d’infertilité, de lésions rénales, hépatiques et du système nerveux central ; - les efforts de l’agence et du laboratoire Pfizer pour empêcher la divulgation des données des essais cliniques ;
- la suppression de rapports de décès ou d’effets secondaires graves dans les bases de pharmacovigilance : plus de 10 000 dans le VAERS américain selon un analyste de données, également dans la base de pharmacovigilance européenne EudraVigilance et celle du Pentagone selon plusieurs médecins ;
- la négation de la supériorité de l’immunité naturelle, pourtant attestée à ce jour par plus de 150 études (Brownstone Institute), au motif qu’elle ne tiendrait pas compte pas du risque de développer une forme grave du COVID en cas d’exposition initiale au virus. Or dans la mesure où les injections favorisent les contaminations et entraînent une aggravation de la maladie en cas d’infection, comme on le sait aujourd’hui, cette objection est en réalité un argument pour ne pas vacciner les personnes déjà immunisées.
- la non-prise en compte des données de biodistribution du vaccin dont on sait depuis plus d’un an qu’il ne reste pas localisé dans le muscle mais circule dans tout le corps et contamine l’ensemble des organes via la circulation sanguine et le circuit lymphatique (Brady et al., Seneff).
Deux éléments remettent notamment en cause la revendication par les laboratoires d’une balance bénéfice-risque positive :
- le risque de perturbation à long terme du système immunitaire, immature chez les tout-petits, si son amorçage initial est biaisé ou qu’il n’est pas optimal, comme le laisse présager les schémas vaccinaux (3 dose) de Pfizer et Moderna.
« Selon la doctrine du péché antigénique originel du Dr Thomas Francis, l’amorçage initial du système immunitaire (exposition initiale au virus, soit dans la nature, soit via un vaccin) est “fixé” à vie. Si l’amorçage initial du système immunitaire est sous-optimal et biaisé, alors cet amorçage initial sous-optimal peut effectivement déranger et biaiser la réponse immunitaire à long terme, ce qui guiderait toutes les réponses immunologiques futures, a déclaré le Dr Paul Elias Alexander, un expert mondial sur le COVID-19. »
Reshaw M. Les conseillers de la FDA approuvent à l’unanimité les injections de vaccins anti-COVID de Pfizer et Moderna COVID pour les nourrissons et les jeunes enfants, ignorant les plaidoyers pour « Ne pas faire de mal ». The Defender. 2022 Jun 15.
- le risque de réponse hyperimmune, potentiellement fatale, induite lorsqu’on vaccine une personne immunisée, ce qui, selon les propres données du CDC, serait le cas de 74,2% des enfants. Comme l’avait déjà expliqué le Dr Urso devant l’Assemblée générale du Tennessee (37’ 30’’), il y a plusieurs mois, c’est précisément en raison de ce risque que Pfizer et Moderna auraient exclu des essais cliniques les personnes qui étaient naturellement immunisées.
On rappellera que c’est pourtant la recommandation officielle des CDC et qu’en France Olivier Véran a toujours refusé de « gracier » les personnes déjà immunisées auxquelles il a au mieux proposé un allègement de peine vaccinale.
Essai clinique Pfizer : ce que montrent les données d’efficacité
L’objectif de la réunion du 15 juin était de présenter les données des essais cliniques conduits par Pfizer et Moderna. Les inquiétudes exprimées par les intervenants extérieurs au comité consultatif de la FDA ont-elles été levées à l’issue de cette audience ? Non selon Toby Rogers, docteur en économie politique spécialisé dans la réglementation des substances toxiques, qui a détaillé les principaux résultats de l’essai clinique Pfizer.
Les défauts de conception de l’étude rendent ses résultats inutilisables
L’essai clinique (no C4591007) a débuté le 24 mars 2021 (fin de la phase 1 : le 16 juillet 2021) pour une date de fin fixée au 14 juin 2024. L’étude était supposée consister en un essai en double randomisé en double aveugle (ventilation aléatoire des participants entre le groupe vacciné et le groupe placebo, à l’insu de l’examinateur et du participant, aucun ne sachant donc ce qui lui est injecté), conduit sur un total de 4 526 enfants âgés de 6 mois à 4 ans, répartis en deux groupes (p. 5).
Or le protocole a été modifié à trois reprises au cours de l’essai, comme cela avait déjà été constaté dans l’essai adulte :
- le 28 septembre 2021, il a été transformé en essai en simple aveugle sans que Pfizer ne fournisse d’explication (p. 23) ;
- le 3 novembre 2021, Pfizer a commencé à vacciner les participants du groupe placebo, rendant de fait impossible tout suivi de l’innocuité à long terme (p. 23) ;
- le 17 décembre 2021, Pfizer a modifié le schéma vaccinal des participants pour y inclure une troisième dose à partir du 1er février en constatant mi-décembre que le schéma à deux doses ne permettait pas d’obtenir une efficacité suffisante chez les enfants âgés entre 2 et 5 ans :
« Par rapport à la population de 16 à 25 ans dans laquelle une efficacité élevée a été démontrée, la non-infériorité a été rencontrée pour la population de 6 à 24 mois mais pas pour la population de 2 à 5 ans dans cette analyse. La décision d’évaluer une troisième dose de 3 µg pour les enfants de 6 mois à moins de 5 ans reflète l’engagement des entreprises à sélectionner avec soin la bonne dose pour maximiser le profil de bénéfice-risque. Si l’étude à trois doses réussit, Pfizer et BioNTech prévoient de soumettre des données aux régulateurs pour soutenir une autorisation d’utilisation d’urgence (EUA) pour les enfants de 6 mois à moins de 5 ans au cours du premier semestre 2022. »
Pfizer. Pfizer et BioNTech font le point sur les études en cours sur le vaccin COVID-19. 2021 Dec 17.
Les résultats sont dès lors impossibles à interpréter, cette évolution du protocole permettant à Pfizer de jouer sur les différentes tailles d’échantillon en fonction des besoins pour « démontrer » les bénéfices ou les risques de son vaccin.
Le mode de calcul de l’efficacité est totalement aberrant
Plusieurs critères permettent d’évaluer l’efficacité d’un vaccin : différence entre le nombre de cas observés dans les groupes vacciné et placebo lors de l’essai clinique, niveaux d’anticorps, nombre nécessaire d’individus à vacciner (NNTV), ce dernir calcul étant théoriquement exigé des laboratoires aux termes de la loi.
Comme nous l’expliquions dans un précédent article, les risques de COVID-19 sont si faibles dans la population infantile, que le nombre nécessaire d’enfants à vacciner pour prévenir un seul cas de COVID est infini. Le calcul du NNTV n’a donc été exigé ni de Pfizer ni de Moderna puisqu’aucun décès d’enfant n’est intervenu ni dans leurs essais cliniques respectifs.
Comment ont-ils alors mesuré la protection conférée par leur vaccin ? Selon The Defender, l’efficacité contre l’infection a été calculée en évaluant la présence d’anticorps, mais cette évaluation n’a aucune valeur scientifique dans la mesure où :
- elle a été réalisée à partir de seulement 225 échantillons de sang, soit un peu moins de 5% de l’effectif initial et sans qu’on sache comment les participants ont été sélectionnés ;
- aucun prélèvement de sang n’a été réalisé dans le groupe placebo. La comparaison a donc été réalisée avec une sélection de 170 échantillons de sang de personnes âgées entre 16 et 25 ans, évaluées lors d’un précédent essai, sans aucune explication sur la manière dont ces échantillons ont été choisis ;
- aucun « corrélat de protection » n’a été mis en évidence : on ne peut donc pas prédire qui sera immunisé.
L’efficacité du vaccin contre l’infection est négative entre la dose 1 et la dose 2 : –30% en moyenne
Pour compléter cette analyse, Pfizer a soumis au comité consultatif une analyse d’efficacité post hoc consistant à comparer les cas de COVID-19 survenus pendant l’essai clinique dans chacun des groupes de participants et pour chaque tranche d’âge, soit 225 dans le groupe vacciné (3013 participants) et 150 dans le groupe placebo (1513 participants).
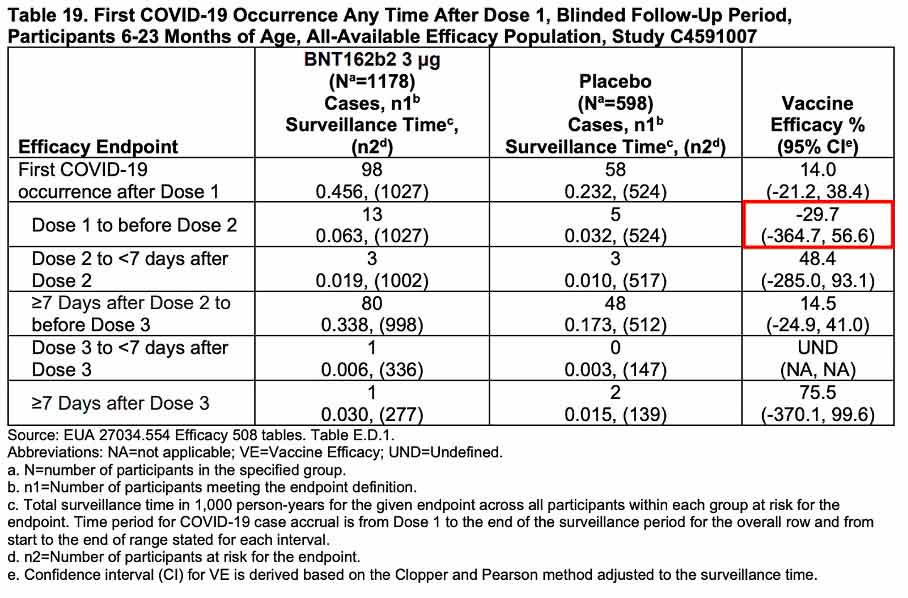
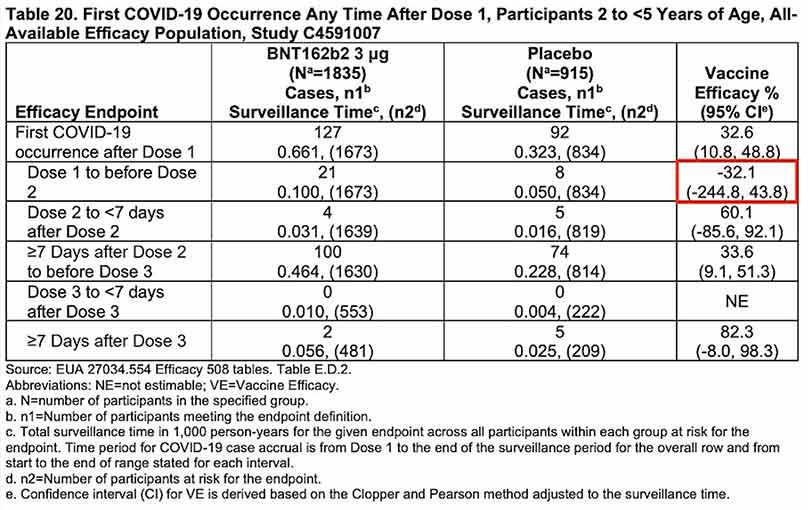
Ils confirment l’immunosuppression induite par le vaccin entre la 1re et la 2e dose dans chacun des deux groupes (efficacité négative de –29,7% entre 6 mois et 2 ans et –32,1% entre 2 et 5 ans), mais surtout, ils sont cohérents avec les données d’observation collectées en population générale et les résultats de l’essai clinique réalisé dans la population adulte sur les précédentes souches du SARS-CoV-2, beaucoup plus virulentes qu’Omicron :
- effondrement immunitaire dans les 7 premiers jours suivant la 1re injection ;
- efficacité positive pendant quelques mois seulement après la 2e dose ;
- érosion progressive de l’efficacité qui devient nulle à 6 mois (Nordström et al./Lancet), négative à compter du 7e mois.
Les dernières données d’efficacité rapportées sont encore plus catastrophiques puisque :
- Chez les personnes ayant été injectées 4 fois, elle serait désormais négative dès la 4e semaine selon une étude israélienne publiée dans le New England Journal of Medicine.
- Selon une étude conduite par le Département de la santé de l’État de New York entre le 13 décembre 2021 et le 30 janvier 2022 et portant sur plus d’un millions d’enfants, l’efficacité du vaccin Pfizer s’établirait aujourd’hui à 12 % chez les enfants de 5 à 11 ans.
Le Dr Clare Craig, pathologiste, coprésidente du groupe Hart, alerte également sur l’existence (p. 38) de 12 enfants ayant contracté deux fois le COVID durant l’essai, dont 11 étaient des enfants vaccinés, la plupart du temps 3 fois.
L’efficacité vaccinale oscillait entre 14 et 60% avant la 3e dose
Le vaccin multiplie le risque de forme grave par 3
Pfizer a au contraire présenté l’efficacité de son vaccin de la manière de la suivante : 10 cas d’infection survenus dans les 7 jours suivant l’injection (en l’occurrence la 3e), dont 7 dans le groupe placebo versus 3 parmi les enfants vaccinés. Or elle ne tient pas compte :
- des risques de contamination induits par l’immunosuppression initiale et l’absence d’efficacité avant la 3e dose, qui oscillait entre 14 et 60% dans l’essai clinique. Ainsi, Pfizer a omis 365 cas sur 375, soit 97% des cas, dont 317 survenus entre les doses 2 et 3 (85%) ;
- des risques vaccinaux contre lesquels l’immunité naturelle protège à 100% ;
- des 8 formes sévères de COVID survenues au cours de l’essai, dont 7 dans la tranche d’âge 2-5 ans, et qui étaient ventilés de la manière suivante : 6 dans le groupe vacciné versus 1 dans le groupe placebo.
Dans la mesure où il y a deux fois moins de participants dans le groupe placebo, le risque de formes graves est multiplié par 3 dans le groupe vacciné.
Détail qui a son importance, les critères de forme grave étaient une augmentation de la fréquence cardiaque ou respiratoire. On peut donc légitimement se demander si cette catégorisation est une manière pudique de dissimuler la cardiotoxicité des vaccins, largement documentée à ce jour chez les enfants et les jeunes adultes ?
Le nombre de cas positifs pourrait être multiplié par 4
L’intervalle de confiance est disproportionné
Un autre élément pousse à remettre en cause les allégations du laboratoire concernant l’efficacité vaccinale : l’intervalle de confiance, c’est-à-dire la marge d’erreur entre les résultats rapportés et l’efficacité réelle. Chez les nourrissons, l’extrémité inférieure a été fixée à –369% pour le calcul de l’efficacité de la 1re dose, –285% pour la 2e dose et, plus inquiétant, –370% pour l’efficacité offerte par la 3e dose. Cela signifie donc que le nombre de cas positifs pourrait être multiplié par 4.
Données de sécurité : des signaux d’alerte majeurs
Le rapport de la FDA fait état de 51 événements indésirables graves (EIG), dont 7 (6 dans le groupe vacciné) ayant provoqué le retrait de l’étude. Ils étaient répartis de la manière suivante (p. 51) :
- entre 6 mois et 2 ans : 17 EIG dans le groupe vacciné, 14 dans le groupe placebo ;
- entre 2 et 5 ans : 12 EIG dans le groupe vacciné, 8 dans le groupe placebo.
Le lien avec le vaccin a été rejeté pour tous les EIG hormis 1 cas de fièvre et de douleur au mollet, mais le nombre anormalement élevé d’EIG observés dans le groupe placebo interroge :
- quelle était la composition du placebo ? Celle-ci n’a jamais été contrôlée de manière indépendante. S agit-il effectivement d’une solution saline, comme l’affirme Pfizer ou d’un excipient actif, visant à minorer la toxicité du vaccin ?
- pourquoi les enfants ont-ils continué à recevoir les autres vaccins pédiatriques ? La FDA précise que la proportion d’enfants concernés était la même dans le groupe placebo et le groupe vacciné (p. 28), mais ce choix de ne pas interrompre le programme de vaccination pédiatrique constitue un facteur de confusion majeur puisqu’il n’est pas possible en l’état de savoir à quel vaccin les EIG sont imputables dans le groupe vacciné.
Enfin – et cette question est peut-être la plus dérangeante –, bien que la fin de l’essai clinique soit fixée au 14 juin 2024, la période de surveillance après l’injection a été limitée à 7 jours. Pourquoi ?
Les événements indésirables graves les plus fréquents étaient les hémorragies menaçant le pronostic vital, le choc anaphylactique, le syndrome anticholinergique, l’encéphalite, l’hypoglycémie et le syndrome des neuroleptiques
Les données de la pharmacovigilance américaine
Compte tenu de ces données, peut-on imaginer que la balance bénéfice-risque soit positive pour les nourrissons et les jeunes enfants ? Pfizer semble en tout cas s’être donné beaucoup de mal pour susciter une efficacité imaginaire et dissimuler des effets secondaires inavouables. Mais au-delà de ces pratiques qui contreviennent à toutes les règles de l’éthique médicale, c’est la démission et la collusion des agences sanitaires qui laissent perplexe aujourd’hui.
La généticienne Alexandra Henrion-Caude, ancienne directrice de recherche à l’Inserm, fondatrice et directrice de l’institut de recherche Simplissima, l’une des plus grandes spécialistes de l’ARN messager alertait il y a quelques jours en ce sens. Elle signale le recensement dans le VAERS (États-Unis) de 58 cas de bébés âgés entre 0 et 3 ans ayant souffert d’effets indésirables post injection, mettant en jeu leur pronostic vital, possiblement jusqu’au décès.
Ces effets indésirables graves sont documentés dans un article publié par une équipe de chercheurs israéliens (Feinberg et Shir-Raz) qui s’interrogent sur l’intégrité des conclusions du comité consultatif de la FDA :
« Les événements indésirables graves les plus fréquents étaient les hémorragies menaçant le pronostic vital, le choc anaphylactique, le syndrome anticholinergique, l’encéphalite, l’hypoglycémie et le syndrome des neuroleptiques. Dans la plupart des cas signalés, il s’agit de blessures multisystémiques. Dans certains cas, on ne sait pas ce qui est arrivé aux bébés : ont-ils survécu ? Et si oui, ont-ils récupéré ? La plupart des rapports ne précisent pas dans quelles circonstances les nourrissons ont été vaccinés et s’ils ont participé aux essais cliniques. »
Feinberg R, Shir-Raz Y. 58 bébés qui ont reçu des vaccins à ARNm COVID-19 ont subi des effets indésirables potentiellement mortels. RT Mag. 2022 Jun 13.
Les auteurs citent plusieurs rapports d’événements indésirables, tous survenus après une injection du vaccin Pfizer-BioNTech contre le COVID-19. Fait particulièrement choquant, parmi ces 58 bébés, 28 avaient moins de 6 mois, ce qui signifie qu’ils ont été vaccinés hors de tout cadre légal puisqu’aucune autorisation de mise sur le marché n’a été délivrée à ce jour pour cette tranche d’âge.
• Arrêt cardiaque chez un bébé de 2 mois, survenu 1 heure après l’injection suivie d’une surveillance de 15 minutes (ID VAERS : 1015467, 02 février 2021). Le rapport précise « Douleur thoracique ; Arrêt cardiaque ; Peau froide et moite » mais ne permet pas de savoir dans quelles conditions cet enfant a été vacciné ni quelle a été l’issue de cet événement.
• Réaction anaphylactique chez une petite fille de 43 jours, suivie de plusieurs blessures multisystémiques engageant le pronostic vital : « Asthme/bronchospasme (étroit) ; Syndrome anticholinergique (large) ; Dépression respiratoire centrale aiguë (large) ; Hypertension pulmonaire (large) ; Cardiomyopathie (large) ; Pneumonie à éosinophiles (large) ; Troubles vestibulaires (large) ; Hypersensibilité (large) ; Insuffisance respiratoire (étroit) ; Réaction médicamenteuse avec éosinophilie et syndrome de symptômes systémiques (large) » (ID VAERS : 1133837, 30 janvier 2021). Le rapport indique que l’enfant n’est pas décédé mais qu’il n’était pas rétabli lorsque le dossier a été enregistré. On ne sait pas si elle est vivante ou non aujourd’hui.
• Choc anaphylactique chez un bébé de 6 mois suivi de plusieurs infections et symptômes multisystémiques : « Syndrome anticholinergique ; Syndrome neuroleptique ; Pneumonie infectieuse ; Autres infections et symptômes multisystémiques » (ID VAERS : 2084418, 29 décembre 2021). Résultat inconnu.
• Syndrome de Guillain-Barré chez un bébé de 1 an : « Syndrome de Guillain-Barré ; Paralysie faciale ; Encéphalite non infectieuse ; Méningite non infectieuse ; Maux d’oreille ; Troubles auditifs » (ID VAERS : 1012508, 19 janvier 2021). Le rapport précise que la vaccination est intervenue hors d’un essai clinique, sans suivi possible.
• Saignements vaginaux chez un bébé de 1 mois, survenus le lendemain de la vaccination : « Saignements vaginaux/saignements vaginaux abondants constants avec des morceaux de caillot » (ID VAERS : 1379484, 19 mai 2021). Là encore, l’issue est inconnue, mais aucune information n’est attendue.
Feinberg R, Shir-Raz Y. 58 bébés qui ont reçu des vaccins à ARNm COVID-19 ont subi des effets indésirables potentiellement mortels. RT Mag. 2022 Jun 13.
La FDA vient d’ouvrir une boîte de Pandore en faisant sauter cette ultime digue, et chacun sait aujourd’hui qu’elle ne le fait pas au nom de la science et de l’intérêt des enfants
La FDA pouvait-elle décemment ignorer ces cas ? Il est difficile de le croire, a fortiori s’ils sont intervenus dans le cadre d’un essai clinique. Mais quand bien même le régulateur américain n’aurait pas couvert ces « vaccinations sauvages », potentiellement fatales pour des bébés dont il sait pertinemment que le risque de décéder de ces injections est supérieur à celui de mourir du COVID-19, rien ne saurait justifier le fait qu’il aide aujourd’hui les laboratoires à maquiller leurs propres données cliniques.
Car si l’objectif revendiqué par le comité consultatif est de laisser la possibilité aux parents qui le souhaiteraient de protéger leur enfant contre le COVID-19, exigeraient-ils de pouvoir avoir excès aux vaccins s’ils en connaissaient la balance bénéfice-risque ? Quelle garantie est aujourd’hui offerte à tous les autres parents que ces injections ne seront pas demain obligatoires ?
La FDA vient d’ouvrir une boîte de Pandore en faisant sauter cette ultime digue, et chacun sait aujourd’hui qu’elle ne le fait pas au nom de la science et de l’intérêt des enfants.
L’enjeu de l’homologation du vaccin pédiatrique : offrir un bouclier juridique aux laboratoires
Peut-on parler d’un « pacte de corruption » ? Factuellement oui, selon les propres termes du directeur général de la FDA, filmé ici à son insu, et dont les révélations ont été confirmées par un document publié par l’agence elle-même, faisant état d’un virement reçu du laboratoire Pfizer pour accélérer l’homologation de son vaccin contre le COVID-19. Détail singulier, cet ordre de virement a été adressé à la directrice du Bureau de la recherche et de l’examen des vaccins à la FDA (Marion Gruber) dont le mari n’est autre que le responsable du développement clinique mondial des vaccins Pfizer (William C. Gruber). En droit français, on appelle ça une rétro-commission. Business as usual.
La corruption ne suffit toutefois probablement pas à expliquer un tel reniement de la science, mais la raison plus obscure invoquée par l’avocat Robert F. Kennedy, Jr engagé dans la défense de victimes présumées des injections confirme la criminalité de cette démarche que l’on peut qualifier de « sordide » : la vaccination pédiatrique est la mère de toutes les batailles puisqu’aux termes de la législation américaine, une fois qu’un médicament est autorisé pour un usage pédiatrique, son fabricant bénéficie d’une immunité absolue contre d’éventuelles poursuites judiciaires.
Le projet Future Framework : l’enterrement de la science
Pour ceux qui doutent encore de la détermination absolue de Pfizer et Moderna à injecter un produit qu’ils savent toxique (la manipulation de leurs données cliniques et l’immunité judiciaire totale qu’ils ont négociée avec les États), cette information révélée par Children’s Health Defense contribuera peut-être à leur ouvrir les yeux :
« Pfizer et Moderna ont un problème : leurs injections COVID-19 ne fonctionnent pas. Tout le monde le sait. Les injections n’arrêtent pas l’infection, la transmission, l’hospitalisation ou la mort. […] C’est un problème parce que les injections reformulées signifient de nouveaux essais cliniques et de nouveaux examens réglementaires par la FDA des États-Unis. Il y a une chance décente que tout vaccin reformulé échoue à un nouvel essai clinique et le public est profondément sceptique à l’égard de ces vaccins, de sorte que l’examen minutieux serait intense. Pfizer et Moderna ont donc trouvé un moyen d’utiliser la capture réglementaire pour faire approuver leurs injections COVID-19 reformulées SANS autres essais cliniques. Leur programme s’appelle le Future Framework et il sera voté par le Comité consultatif sur les vaccins et les produits biologiques apparentés (VRBPAC) de la FDA le 28 juin. »
Rogers T. La FDA autorisera-t-elle Pfizer et Moderna à ignorer les essais cliniques pour les futurs vaccins COVID ? The Defender. 2022 Jun 01.
L’astuce ? Assimiler le vaccin contre le COVID à celui contre la grippe et lui appliquer le processus de sélection des souches grippales. C’est ce processus qui permet, chaque année, d’élaborer un vaccin contre le variant en circulation, sans avoir à conduire de nouvel essai clinique. Tous les futurs vaccins contre le COVID-19, quelle que soit leur formulation, seront alors « automatiquement considérés comme “sûrs et efficaces” sans essais cliniques supplémentaires, car ils seront considérés comme “biologiquement similaires” aux vaccins existants » (The Defender).
Problème, la nouvelle technologie vaccinale à ARN messager, utilisée dans les vaccins contre le COVID-19 n’est ni stabilisée ni même homologuée, la date de clôture de l’essai clinique n’étant prévue qu’en 2024 pour Pfizer (nous avons détaillé les dates de clôture pour les autres vaccins dans un précédent article). Second problème, même si une étude conduite cette année par l’Université du Michigan a révélé que l’efficacité du vaccin contre la grippe était « littéralement nulle » (les auteurs ont calculé une efficacié vaccinale égale à 0), il est a minima beaucoup moins toxique que les vaccins à ARN messager (Montano) :
« La dyspnée, l’arrêt respiratoire, l’embolie pulmonaire, l’infarctus du myocarde, la thrombose, les hémorragies cérébrales et la pneumonie étaient les effets indésirables les plus fréquemment mentionnés dans les rapports de décès. […] Il existe un important excès de risque de décès, d’hospitalisation et de rapports mettant en jeu le pronostic vital pour tous les vaccins COVID-19 par rapport aux vaccins antigrippaux. »
Motano D. Frequency and Associations of Adverse Reactions of COVID-19 Vaccines Reported to Pharmacovigilance Systems in the European Union and the United States. Front Pub Health. 2022 Feb 03. DOI: 10.3389/fpubh.2021.756633.
Si ce projet de Future Framework est validé par le comité consultatif de la FDA qui a considéré à l’unanimité que la balance bénéfice-risque des vaccins pédiatriques Pfizer et Moderna était positive, il n’y aura donc plus de déshonneur à prescrire un poison sur ordonnance, juste à fermer les yeux et laisser les laboratoires injecter ce qu’ils désireront, autant de fois qu’ils le souhaiteront, selon le business plan qu’ils auront établi.
